| This is an old, no longer maintained version of the website, for new one please visit: http://gsn.io.gliwice.pl |

 |
 PROTEIN STRUCTURE PREDICTION BY CONSENSUS FOLD RECOGNITION AND ASSEMBLY OF FRAGMENTS Janusz M. Bujnicki Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology, Trojdena 4, 02-109 Warsaw, Poland e-mail: , web: http://genesilico.pl In the age of structural genomics, the computational assignment of three-dimensional structures to newly determined protein sequences is becoming an increasingly important complement to experimental structure determination. In particular, fold-recognition methods aim to build 3D models for proteins bearing no evident sequence similarity to any protein of known structure by identification of structural "templates" compatible with the modeled sequence. Meta-servers run a variety of primary methods to perform the fold-recognition analysis, collect and compare the results, and select one of the models as the most representative. On the average, this approach leads to much better predictions of protein structure than using any single "primary server". Recently, a new generation of prediction methods appeared, which build hybrid models from fragments, either obtained from the experimentally solved protein structures, or from the fold-recognition models. The general premise of this approach is that the protein conformation is reasonably well approximated by the distribution of local structures adopted by known, not necessarily homologous, protein structures. The "fragment splicing" methods are very powerful, as the resulting hybrid models are on the average more complete and more accurate than the input models. They can also correctly predict the 3D structure of a protein with a completely new fold, which could not be modeled based on any single template. Currently, the accuracy of models built by the fully automated meta-servers and "fragment splicing" methods approaches the models built by best human modelers; however, it still lags behind the quality of high-resolution structures obtained by X-ray crystallography. I will review the state-of-the-art in the field of protein structure prediction, with the emphasis on what is currently possible and what is not (yet) possible. REFERENCES 1. Proteins, Structure, Function, and Genetics: Volume 53, S6, 2003 Supplement 6: CASP 2. Practical Bioinformatics, Janusz M. Bujnicki (ed.), Springer-Verlag, 2004 RELEVANT LINKS http://predictioncenter.llnl.gov/ http://www.genesilico.pl/meta/ http://robetta.bakerlab.org/ RESPONSE TO CHALLENGING DOSE OF X-RAYS AS BIOMARKER OF SUSCEPTIBILITY IN MOLECULAR EPIDEMIOLOGY. Antonina Cebulska-Wasilewska1,2 1 Department of Radiation and Environmental Biology, the H. Niewodniczański Institute of Nuclear Physics, PAN, Kraków, 2 Chair of the Epidemiology and Preventive Medicine, CM UJ, Kraków, Poland The importance of various environmental exposures has been evident in variation in cancer incidence and mortality. Elevated frequencies of chromosome aberrations or sister chromatid exchanges in human lymphocytes are recognisable biomarkers of hazardous genotoxic effects caused by environmental or other exposures. Results of the cytogenetic damage detected in our monitoring studies have shown both an association with an adverse health outcome on one side and the influence of confounding factors related to the lifestyle on the other. The single cell gel electrophoresis assay, the alkaline version in particular, has become a very popular method for the analysis of the DNA damages caused by various chemical and physical agents. Comparison between DNA damage investigated by the SCGE assay and cytogenetic damage induced in human lymphocytes by chemicals or ionising radiation revealed a strong correlation between the two assays. Obtained results suggested that the SCGE assay provides a good prediction of cytogenetic damage, and because of its simplicity and rapidity, the assay would appear to be a useful tool for the predicting the individual susceptibility to the induction of the DNA damage or for determining the genotoxicity of environmental agents. This review is based on studies in which radiation was applied as a challenging dose and DNA damage induced and repaired was analyzed with the use of the single cell gel electrophoresis (SCGE) assay. Results from studies on susceptibilities and repair competence carried out in various groups of exposed workers, controls, and cancer patients (more than 700 donors) show variability between donors both in a response to challenging treatment and in the efficiency of repair process. Influences on cellular capacities of the occupational exposures and other factors depending on genotypes or life style are observed. On the basis of presented results it could be suggested that application of ionizing radiation as a challenging treatment and SCGE as the method for DNA damage measurement in combination with repair competence assessment might be used in a molecular epidemiology or in preclinical studies as a fast biomarker predicting for various donors a cellular susceptibility to various genotoxins and exposures (environmental, occupational or therapeutic). Acknowledgments Research was partially supported by grants: EC: CRB-NAS QLK4-CT-2000-00628 and SPUB No 156/E-390/5.PR UE/DWM 626/2003-2004. NEW TRENDS IN EXPERIMENTAL ONCOLOGY IN UKRAINE V.F.Chekhun R.E.Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology NAS of Ukraine, Kiev, Ukraine Currently, knowledge of tumorigenesis biology does not allow precise diagnosis of the disease based on traditional diagnostics methods as they do not permit characterizing biological peculiarities of the tumor nor detecting micrometastases. Presently, the combination of cytogenetic, immunohistochemical, immunocytochemical and molecular-biological methods, along with classic morphological methods, should be applied thus allowing determination of biological characteristics of the tumor and its metastatic potential. Novel molecular biological technologies that are developed in IEPOR allow detection and correction of pathogenetic alterations in transformed cells via system of antigenic determinants and cytokine spectrum. In particular, use of monoclonal antibodies (MoAbs) allows diagnosing hemoblastosis in cancer patients (adults and children) in 95-98% of cases, to describe 7 variants of acute leukemia and 8 variants of acute myeloid leukemia that forms the basis for modern classification of lymphoproliferative diseases according to WHO requirements. Such precise diagnosis allows more effective therapeutic applications, i.e. to apply conjugates of MoAbs with antitumor drugs. The technology created in IEPOR is based on the use of MoAbs of IEPOR series (IPO-4) that initiates programmed death of tumor cells. Another research direction is the development of methods for immunotherapy of cancer based on construction of antitumor vaccines using substances of microbial origin (i.e. products of bacterial synthesis, receptors etc). The latter cause devitalization of autologic tumor cells, effective stimulation of effector cells thus providing antitumor resistance. Such autovaccines may be used either as additional remedy, or as immunomodulators. In conclusion, one may suppose that the priority in the development of new strategies for treating cancer patients will belong to early diagnosis and individualization of therapy based on the achievements of modern biotechnology. The determination of markers for early diagnosis, production of highly specific cytostatics and vector systems for their transportation, generation of new vaccines, probiotics and MoAbs that directly influence the key chains of cell differentiation and apoptosis will form a way to success in solving problems of clinical oncology. REPAIR OF DNA DOUBLE-STRAND BREAKS OF DIFFERENT ORIGIN IN SACCHAROMYCES CEREVISIAE Miroslav Chovanec1, Viera Vlckova, Eva Markova1, Zuzana Dudasova1, Andrej Dudas1, Danusa Vlasakova1, Katarina Hermanska2, Daniela Gabcova2 and Jela Brozmanova1 1Laboratory of Molecular Genetics, Cancer Research Institute, Vlárska 7, 833 91 Bratislava 37, Slovak Republic and 2Department of Genetics, Comenius University, Faculty of Natural Sciences, 842 15 Bratislava, Slovak Republic DNA double-strand breaks (DSB) are considered the most severe type of DNA damage, because if left unrepaired, they can cause cell death. If misrepaired, they can lead to genomic instability and, ultimately, the development of cancer in multicellular organisms. Two main pathways have evolved in eukaryotic cells for the repair of DSB, homologous recombination (HR) and non-homologous end-joining (NHEJ). DSB can be induced by a variety of agents. To address the question how oxidatively induced DSB are repaired in S. cerevisiae, we have studied survival, mutagenesis and DSB repair in cells debilitated in HR and NHEJ after treatment with hydrogene peroxide (H2O2), bleomycine (BLM) and menadione. Our results indicate that defect in HR or NHEJ increases sensitivity of yeast cells to BLM, but not H2O2 or menadione. Only if HR and NHEJ were impaired simultaneously, yeast cells were also sensitive to H2O2 and menadione. Pulsed-field gel electrophoresis experiments showed no DSB induction in HR- or NHEJ-defective cells after H2O2 or menadione treatment, although after BLM exposure DSB were extensively induced. Therefore, toxicity of H2O2 or menadione does not seem to be primarily caused by DSB induction as is case of BLM exposure. BLM-induced DSB were not repaired in HR-defective cells, suggesting that HR is the main mechanism involved in their repair in yeast. Mutation frequency induced by all oxidising agents was significantly increased only in HR-defective cells, supporting an existing view that DSB repair by HR is error-free process. DSB can also be induced indirectly as intermediates during interstrand cross-link (ICL) repair. In S. cerevisiae, there is a group of mutants that are specifically sensitive to ICL-inducing agents. Of them, pso2, seems to be defective in the repair of DSB that are induced by these agents. Since ICL repair involves the DSB repair proteins to complete the process, we have verified a possibility of interaction between the Pso2 and HR or NHEJ proteins. These results will be discussed. IDENTIFICATION OF POTENTIAL CIS-REGULATORY FEATURES ASSOCIATED WITH A MODE OF GENE EXPRESSION COMMON TO NEURONAL DIFFERENTIATION AND HIPPOCAMPAL DEVELOPMENT M. Dabrowski1, S. Aerts2, Y. Moreau2, and B. Kaminska1 1Nencki Institute, Warsaw, Poland 2Katholieke Universiteit Leuven, Belgium Availability of genomic sequence data of several species and of results of gene profiling from related biological systems permits a comparative approach to analysis of gene regulation. We analysed temporal gene expression profiles from published gene profiling experiments in two related biological systems: developing mouse hippocampus in vivo, and neurons undergoing differentiation in vitro, in conjunction with the genomic sequence of putative cis-regulatory regions, identified by mouse-to-human homology. We sought to identify features of the cis-regulatory regions common to many genes and predictive of particular expression pattern(s). We assumed that cis-regulatory regions of many genes share functional modules that have independent and additive (in log representation) effects on temporal expression profiles. Motivated by the above ("mode-module") hypothesis, before analysis of cis-regulatory regions, we orthogonalized the expression data by the singular value decomposition (SVD). We demonstrate that a particular SVD mode is highly conserved between two experimental systems. The conserved mode reflects, depending on the sign, a component of continuous up-, or continuous down-regulation, in the course of either experiment. Using a candidate features approach, with pairs of motifs representing transcription factor binding sites as the candidates, we identified ten features, all conserved between the mouse and the human, that were predictive of the sign of the conserved mode corresponding to down regulation, when tested on the other half of the data than the half originally used to identify them. Two of the identified features were predictive of down-regulation in both experimental systems (a cross-system test). The relationship between the (unknown) gene expression space, the measurement space, and the subspaces of the SVD modes, will be discussed in view of the mode-module hypothesis. EXPRESSION OF MEIOTIC GENES AND PHENOTYPE BY IRRADIATED TUMOUR CELLS J. Erenpreisa1, M. Kalejs 1, A. Ivanov 2, M. Cragg 2; B. Liebe 3, H. Scherthan3 1Biomedical Centre Latvian University, Riga; 2Cancer Science Division, Southampton; 3Max Planck Institute for Molecular Biology, Berlin. Many malignant tumours are expressing the so called cancer-testes associated genes and explanation for such a selective genome dysregulation is lacking. Our interest in this issue was stimulated by previous findings that radioresistant irradiated lymphoblastoid cell lines produced after mitotic catastrophe polyploid cells, which displayed some features of meiotic prophase and were capable to undergo DNA repair by homologous recombination and resist apoptosis. In this study we investigated in this model the expression of several meiotic genes and associated phenotype. It was shown that a principal meiotic regulator Mos/MEK/MAPK pathway is activated in p53 mutants by irradiation at the peak of arrest in a spindle checkpoint, proportionally to it, to the number of subsequently emerged polyploids, and to clonogenicity. In turn, inhibition of MEK/MAPK prevents catastrophic mitoses, reduces polyploidy and clonogenicity. Meiotic cohesin Rec8 is activated and appear in the structured form in the chromatin of arrested metaphases restituting into polyploid cells. The pattern of Rec8 immunofluorescence in most giant tumour cells corresponds that of preleptotene in the positive rat testes control. Correspondingly, we observed location of telomeres on the nuclear envelope characteristic for leptotene. The central and lateral elements of synaptonemal complexes (SC), SCP1 and SCP3 are transcribed, however at low levels. SCs and serial mitoses within the same giant cells, reducing the DNA content from 16C to 2C, were occasionally observed. Expression of all meiotic genes mentioned above was found in non-treated cells at the very low level. Conclusion: some kind of a meiosis-like program, although abortive in most emerged polyploid cells is activated by mitotic catastrophe in lymphoblastoid p53 mutants. EPIGENETIC REGULATION OF TRANSCRIPTION AND THE ORIGIN OF ISOCHORES Jan Filipski Institut Jacques Monod, Paris, Instytut Biologii Uniwersytetu im. M. Kopernika, Toruń The results obtained by density gradient centrifugation in early 70-es indicated that DNA in eucaryotic genomes (especially in mammals) is compositionally heterogeneous at high molecular weight level. In particular, it was found that the composition of large DNA segments (50 kb long and longer) vary between 35 and 65% GC in human genome. The long genomic regions, which showed some compositional homogeneity, were called isochors (from Greek "equal landscape"). The evolutionary origin of the isochors became understood only recently. The likely explanation suggests that the frequently arising new alleles containing AT base pairs in the GC-rich genomic segments have a short life and are being eliminated from the population in part by selection and in part by a process independent from selection. The process which can eliminate the AT carrying alleles from a population without physically eliminating their carriers could be the biased gene conversion (non crossover recombination) occurring during meiosis. Indeed, the hotspots of recombination in eucaryots coincide with the GC rich DNA segments. These DNA sequences showing increased recombination frequency are located in the euchromatic regions of the chromosomes, in the vicinity of the transcriptional factories in the nucleus, and contain the genes available to transcription. The rarely recombining AT-rich DNA carries the genes located in the heterochromatic chromosomal regions, frequently at the periphery of the nucleus. The transcription of the genes belonging to this class is accompanied by their transportation into the vicinity of the transcriptionally active nuclear sites. This displacement is associated with epigenetic changes of the chromatin fibre carrying the activated genes. ISOTHERMIC SEQUENCING BY HYBRIDIZATION Piotr Formanowicz Reading of DNA sequences remains one of the most important problems in molecular and computational biology. Although human genome has been almost fully sequenced and also genomes of some other species are already known there is a need for an efficient and inexpensive method for DNA sequencing. The two main methods for determining DNA sequences are shotgun sequencing and sequencing by hybridization (SBH). The latter one consists of two phases: the biochemical one and the computational one. At the biochemical stage, hybridization experiment is performed, in which an oligonucleotide library is compared with many copies of one of the strands of the examined DNA molecule. Usually, the library consists of all 4l oligonucleotides of length l. During the hybridization reaction fragments of the target DNA attach to oligonucleotides from the library in their complementary places. After the reaction one obtains a set of sequences being subfragments of the examined DNA molecule. This set is called a spectrum. In the second phase of the method a permutation of spectrum elements corresponding the the target DNA sequence is determined. In the case of an ideal hybridization experiment (i.e. without experimental errors) the target DNA sequence may be reconstructed in polynomial time. On the other hand, if there are errors in the experiment (positive ones - excess of oligonucleotides in spectrum or negative - lack of some oligonucleotides), the computational phase of the method becomes a strongly NP-hard problem. Hence, a challenging problem is to design some new approach reducing a number of errors in the biochemical stage of SBH. Such an approach is isothermic SBH, which is based on the usage of oligonucleotide libraries consisting of oligonucleotides of equal melting temperature, instead of oligonucleotides of equal length. It is known that duplexes of C/G rich oligonucleotides are more stable than A/T rich ones. This phenomenon may result in numerous errors of positive type. At the same time a modification of hybridization conditions directed towards diminishing the number of imperfect duplexes of C/G rich oligonucleotides may result in increasing a number of missing perfect duplexes of A/T rich oligonucleotides. The duplex formation depends on base composition and on its length. Thus, at least formally, it is possible to compensate lower stability of A/T rich duplexes by increasing their length. The key idea of isothermic SBH is to obtain a set of oligonucleotides that differ in base composition and length and are characterized by a predefined relation between base composition and length of oligonucleotides. In a specific case, where an increment of C or G is twice that of A or T and the sum of increments for each oligonucleotide in the set is constant, the set is called isothermic oligonucleotide library. It may be shown that two such libraries are sufficient to reconstruct DNA sequences by SBH approach. IDENTIFICATION OF MODELS OF CELL SIGNALING PATHWAYS Krzysztof Fujarewicz This work concerns the problem of fitting mathematical models of cell signaling pathways to real data. We propose the adjoint sensitivity analysis as a very efficient tool for solving such a problem. Mathematical models of cell signaling pathways frequently take form of a set of nonlinear ordinary differential equations. To compare different models and to test their ability to model processes, for which experimental data are given, an effective method of parameter fitting is needed. Unfortunately, while the model is for time continuum, all available measuring techniques, for example: blotting techniques, electrophoretic mobility shift assays or gene expression microarrays give measurements only at discrete time moments. Adjoint sensitivity analysis is a technique frequently used in practical optimization problems such as identification or optimal control tasks. It calculates a gradient of performance index very effectively and decreases computational costs when compared to straight (tangent linearized) sensitivity analysis. In literature devoted to adjoint systems such systems are defined as continuous in time systems or discrete in time systems and cannot be directly applied to solve the problem of parameter fitting presented above due to its hybrid, continuous-discrete time nature. In this work we present rules for creating the modified adjoint systems for continuous-discrete systems (i.e. systems containing continuous and discrete time parts, pulsers and samplers). Then it is used to solve the problem of finding the gradient of a quadratic performance index, utilized during parameter fitting procedure. The approach is illustrated on mathematical model of NF-ĸB regulatory module. Presented results show very good convergence of solutions in the obtained models to real data. THE FATE OF DNA DOUBLE-STRAND BREAKS: ASPECTS OF MITOTIC AND MEIOTIC CELLS. Wolfgang Goedecke University Duisburg-Essen, Universitätsstraße 5, 45117 Essen, Germany Double-strand-breaks (DSB) represent a major threat to genomic DNA. Possible consequences of these lesions include repair of the DSB, the gain of new mutations in cases of misrepair, cell-cycle arrest and apoptosis. We are interested in exploring how the cell is able to integrate these different physiological responses caused by DSBs. A protein involved in many aspects of DSB processing is MRE11. Initially it has been identified in yeast as a member of the RAD52-epistasis group, required for processing of meiotic DNA-DSB breaks using Homologous-Recombinational-Repair (HRR). In order to find proteins that are involved in the processing of DSBs, a two-hybrid-screen was performed using MRE11 as a bait. Beside expected interactors, for example the spo11-protein, a topo-II like enzyme creating DSBs for the initiation of homologous recombination during meiosis, we have also identified the Ku70-protein, suggesting a role of MRE11 in Nonhomologous-End-Joining (NHEJ). Recently, such a function of MRE11 in by the inhibition of NHEJ-activities in cell-free systems using anti-MRE11 antibodies was found. Obviously, MRE11 is involved in homology-dependent as well as homology-independent DSB-repair mechanisms. The question came up, how MRE11 could participate in such different aspects of DSB-repair. In order to address this question, immunocytology on rat testis sections was used to investigate the localisation of Ku70 and MRE11 in premeiotic and meiotic cells. It turned out, that in cells entering meiosis, the Ku70 protein is no longer detectable. Our current working hypothesis is, that (i) NHEJ is dominant over HRR in mammalian cells and (ii) that the NHEJ activity has to be switched-off during meiosis to allow HRR of the meiotic DSBs to take place. These data are consistent with earlier experiments in extracts from oocytes and eggs of the clawed frog X. laevis, where we could show, that NHEJ activity indeed becomes lost during egg-maturation. PCR AMPLIFICATION OF HIGHLY GC-RICH TEMPLATES. AN EFFICIENT TOOL IN THE SEARCH FOR NEW PROGNOSTIC MARKERS IN CANCER. Lise Lotte Hansen Centre of Molecular Gerontology, Department of Human Genetics, The Bartholin building, University of Aarhus, DK-8000 Aarhus, Denmark e-mail: lotte@humgen.au.dk In the search for new molecular genetic markers with a statistical significant correlation to prognosis in cancer, it is often necessary to use large scale PCR amplification of a large variety of DNA sequences. DNA templates with a high GC content or multiple consecutive Simple Tandem Repeats (STRs) are difficult to handle using a majority of PCR related techniques and can obstruct PCR dependent analyses in the following situations: middot; Standard PCR amplification. Highly GC-rich regions prevent template denaturation and thereby product synthesis. middot; Multiplex PCR amplification. A preferential amplification of low GC-content sequences results in misinterpretation of the balance between the two sequences. middot; Quantitative PCR amplification. GC-rich targets may form heteroduplexes with internal standards, which are designed to be very similar to the target, leading to misinterpretation of the results. middot; DNA sequencing. DNA polymerase tends to stall in GC-rich regions, causing decompressing of the sequence thereby making it impossible to read. middot; cDNA synthesis of GC-rich templates may create shorter fragments that lack GC-rich portions of the sequence. The result is a skewed representation of the original mRNA molecule. middot; Microarray (Not PCR related). The amino acid analogue betaine (N,N,N,-trimethylglycine) has proved very successful as an enhancer of PCR amplification of highly GC rich regions. Betaine is more efficient than other substances among low-molecular weight sulfones as DMSO and tetramethylene sulfone. For some amplifications only betaine is successful as an additive. Reference: Hansen, L.L. and Justesen, J. PCR amplification of highly GC-rich regions. PCR Primer: A Laboratory Manual, Second edition, chapter 5. Edited by Diffenbach, C.W. and Dveksler, G.S.New York: Cold Spring Harbor Laboratory Press. 2003 THE SPATIAL ORGANIZATION OF DNA IN EUKARYOTIC CELL NUCLEI PLAYS AN IMPORTANT ROLE IN DETERMINING POSITIONS OF RECOMBINATION HOT-SPOTS O. V. Iarovaia and S. V. Razin Laboratory of Structural and Functional Organization of Chromosomes, Institute of Gene Biology RAS, Vavilov street 34/5, 119334 Moscow, Russia We have previously found that the major recombination hot-spot of the human dystrophin gene is localized to the nuclear matrix (Iarovaia et al., 2004, Nucleic Acids Res. 32, 2079-2086). In the present study we have analysed the spatial organization in interphase nuclei of the breakpoint cluster regions (bcrs) of the AML-1 and ETO genes frequently participating in reciprocal t(8;21) translocations. Both bcrs were found to be localized preferentially, but not exclusively, to the nuclear matrix, as shown by hybridization of specific probes with nuclear halos. This association was not related to transcription as the transcribed regions of both genes located far from bcrs were located preferentially in loop DNA, as shown by in situ hybridization. The sites of association with the nuclear matrix of the intensely transcribed AML-1 gene were mapped also using the biochemical PCR-based approach. Only the bcr was found to be associated with the nuclear matrix while the other transcribed regions of this gene turned out to be positioned randomly in respect to the nuclear matrix. The data are discussed in the framework of the hypothesis postulating that the nuclear matrix plays an important role in determining the positions of recombination-prone areas, and that inhibition of DNA topoisomerase II of the nuclear matrix may trigger a chain of events resulting in illegitimate recombination. FIDELITY OR SURVIVAL? HOW UNREPAIRED DNA IS REPLICATED WHEN POLYMERASES ENCOUNTER PROBLEMS. Paweł Jałoszyński Institute of Human Genetics, Polish Academy of Sciences, Poznań Recent studies on the translesion DNA synthesis provided much interesting data on particular role of non-replicative DNA polymerases in mutagenic events. Enzymes belonging to the Y-superfamily of polymerases (i.e. pol. eta, kappa, iota) were found to replicate past various DNA base modifications with efficiency and fidelity strongly depending on the type of lesion. Such enzymes are proposed to participate in the "two polymerases affair" action, which assumes a dissociation of replicative (i.e. pol. alpha, delta, epsilon) or repair (i.e. pol. beta) polymerase from damaged template and continuation of the reaction by less sensitive (and often more mutagenic) translesion polymerases. Our study focused on the role of 8-oxoguanine (8-OH-G) in mutagenesis occurring during DNA synthesis catalyzed by polymerase alpha, beta, eta and kappa. To measure kinetic parameters of nucleotide incorporation, we used synthetic fragments of human and mouse ras genes containing one or two 8-OH-G moieties in the hypermutagenic codon 12. Mutations in this particular codon are responsible for permanent activation of the gene, leading to a neoplastic transformation, and the sequence is widely used in translesion DNA synthesis studies. Polymerase alpha was found to be extremely sensitive to 8-OH-G, being strongly inhibited by a single lesion and completely blocked by two oxidized bases in a tandem arrangement; it was able to misreplicate with low efficiency, misincorporating AMP. Polymerase beta was also remarkably inhibited, but not blocked by 8-OH-G, misincorporating AMP opposite the lesion. Polymerase kappa, also inhibited by oxidized guanine, appeared to be most mutagenic in the system applied; the efficiency of misinserting AMP opposite 8-OH-G was highest amongst the studied enzymes. Pol. kappa showed also an interesting effect of "action-at-a-distance" mutagenesis, misinserting AMP opposite non-modified guanine 3'-flanked by 8-OH-G. Polymerase eta, in turn, appeared to be completely insensitive to inhibition by the lesion, even by two 8-OH-Gs in a tandem arrangement. Moreover, this enzyme misincorporated AMP opposite the lesion and (with low efficiency) opposite G 3'-flanked by 8-OH-G. Interestingly, two lesions in a tandem arrangement strongly relaxed the enzyme specificity; in this situation all four dNMPs were incorporated. This unique property may be responsible for increased mutation rates when pol. eta replaces replicative or repair polymarases, which are strongly inhibited or blocked. This action may ensure survival of cells with massive DNA damage, but it leads to dramatically increased mutagenicity. GENOMIC APPROACH TO THYROID CANCER Barbara Jarząb Department of Nuclear Medicine and Endocrine Oncology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Gliwice Branch Gene expression profiling is valuable both looking for multigene classsifiers as potent diagnostic tools in oncology and dividing tumor histotypes into new subclasses, which may fit better to cancer clinics, therapy and outcome. In this way microarray-based analysis may outperform traditional histopathology, paralelly supplying information about the mechanisms of neoplastic transformation and cancer progression. Until now we have analyzed over 70 thyroid tumors and nearly 40 benign thyroid tissues by high density oligonucleotide DNA microarrays (U133 GeneChip, Affymetrix). These results will be presented in comparison to other datasets available in the public domain to address the questions related mainly to papillary thyroid ca (the most frequent form of thyroid cancer). Sources of variance in gene expression profile, its relation to the most important clinical and pathological prognostic features of cancer disease as well as gene expression signature for diagnostic purposes will be addressed The question of the major sources of variance in gene expression profiles of papillary thyroid carcinoma (PTC) as compared to non-tumor thyroid tissue obtained in the same patients was analyzed by Singular Value Decomposition. SVD confirmed the prevalent role of tumor/normal difference and revealed that immunity-related gene expression varied significantly between tissues and constituted the strongest confounding signal (chemokines, T cell receptor genes etc within the 2nd mode and immunoglobulin transcripts prevailing in the 3rd mode). Inter-individual variability played a minor role in our samples. Relations between PTC gene expression profile and clinical and pathological features were analyzed by SAM (Significant Analysis of Microarrays), then by other techniques. Clinical factors related to stage and course of the disease (lymph node metastases, presence of distant metastases or quick relapse) revealed much closer relation to the gene expression profile than classical pathological factors of poor prognosis (tumor diameter, multifocality, capsule infiltration, angioinvasion). By means of a Support Vector Machine-based method developed at the Silesian University of Technology (Recurrent Feature Replacement) a 20-gene classifier was selected to support PTC diagnosis. The validation step was performed on 50 thyroid samples.The classifier correctly differentiated PTC and non-malignant thyroid tissues in 90% of cases. The analysis of misclassified samples showed a subgroup of PTCs which exhibited different expression profile than the majority of tumors. This implies a necessity to include in a classifier a wider variety of gene expression profiles and raises the question of the clinical relevance of the putative PTC subtypes. FROM MICROARRAY DATA TO THE BIOLOGICAL RELEVANCE Michał Jarząb Department of Tumor Biology, Maria Skłodowska-Curie Memorial Institute of Oncology and Cancer Center, Gliwice Branch Vast amount of information, delivered by microarray-based studies, requires special methodology for adequate analysis. Until now the main attention has been devoted to the development of algorithms appropriate for biostatistical and bioinformatical evaluation of these data (preprocessing, multiple testing-related problems, redundancy, multivariate analysis, machine learning, etc.). However, the question how to extract the biological information from the results of microarray studies is still a big challenge for the biomedical community. Some approaches to extract biologically-relevant information from the microarray data will be described and discussed. All presented problems will be discussed in the context of microarray data obtained in our laboratory. Gene ontologies - formal classification systems of various gene and protein properties - form today a key to analysis of large groups of genes. The best known ontology, developed by Gene Ontology Consortium and used by Affymetrix to annotate probes, classifies more than 10,000 human genes. Analysis of the lists of selected "significant" genes is based on classifying them according to their ontology and comparing the number of genes present in the list to the whole class. The most overrepresented ontology classes are then considered relevant from the biological point of view. In my presentation I will show an alternative and simple approach to this task. Before the bioinformatical analysis, the whole dataset is divided according to selected ontology classes. Then, gene selection algorithms are applied to each class separately and the contents and the power of classification are evaluated for each obtained list separately. This approach allows a direct comparison of the relevance of functional gene classes to the problem of interest. The other important point is the comparison of gene expression in obtained lists with publicly available microarray data. Visualizing gene expression in other tissues and other pathologic conditions helps to understand the role of selected genes. The public domain microarray data are growing rapidly and the role of this approach will probably increase in future. MOLECULAR MECHANISMS OF THE INDIVIDUAL RADIOSENSITIVITY: IMPACT OF DNA REPAIR U. Kasten-Pisula, K. Borgmann, I. Brammer, M. Purschke and E. Dikomey Section of Radiobiology and Experimental Radiooncology, University-Hospital Hamburg-Eppendorf, Germany The extent of the normal tissue reaction of patients after radiotherapy show huge variations. The molecular reasons for this individual radiosensitivity are not clarified in detail. So far, it is known that the cellular capacity to repair DNA double-strand breaks (dsbs) affects radiosensitivity, as the number of residual dsbs and the resultant lethal chromosome aberrations correlate with cell survival of human fibroblasts. Furthermore, it was shown that most of all dsbs induced (95-98%) are repaired and that very small variations in dsb repair capacity of only a few percent are sufficient to result in a clear change of cell survival (factor about 2). Therefore, it was of interest to test the impact of dsb repair proteins, especially that of the non-homologous end-joining (Ku70, Ku80, DNA-PKcs, XRCC4), which represents the main dsb repair pathway in mammalian cells. For these experiments different human fibroblasts with clear differences in radiosensitivity (SF3.5Gy: 0.03 - 0.28) were investigated. It could be demonstrated that neither the extent of repair gene / protein expression nor of the DNA-PK activity correlate with cell survival. This indicates that the dsb repair capacity, and also the extent of radiosensitivity, is defined by more complex mechanisms. There are now additional data indicating that also secondary dsbs arising during the repair of clustered base damage might affect cell survival. It could be demonstrated that the repair kinetics of radiation-induced oxidative base damage (8OH-guanine) is clearly faster in rodent compared to human cells (t1/2 20min vs t1/2 80min) and that this corresponds with cellular radiosensitivity (D0.1 rodent 5.2Gy vs human 2.7Gy). Further, a stimulating effect of Tp53 on the repair kinetics of oxidative base damage was shown in Tp53 proficient mouse embryonic fibroblasts (t1/2 21min) compared to Tp53 deficient fibroblasts (t1/2 49min), which again corresponds to the radiosensitivity (D0.1 Tp53 / 6.6Gy vs Tp53-/- 4.8Gy). These data indicate that not only directly induced dsbs but also base damage (cluster) and their repair kinetics might play a role in defining cellular radiosensitivity. GENE EXPRESSION PROFILING IN HEREDITARY BREAST CANCER; PRELIMINARY RESULTS. K. Lisowska1, O. Dudaladava1, M. Jarząb1, J. Pamuła1, W. Pękala1, T. Huzarski2, J. Lubiński2, E. Grzybowska1 1Department of Tumor Biology, Maria Skłodowska-Curie Memorial Cancer Center and Institute of Oncology, Gliwice Branch; 2Pomeranian Medical Academy, Szczecin Gene expression profiling allows better understanding of cancer biology and offers potential practical clinical applications. DNA microarray analyses have been successful in finding potential new molecular markers as well as molecular signatures characteristic for cases with different clinical and pathological properties. Promising findings were achieved e.g. in studies of lymphoma, breast and prostate cancer where distinct sets of genes were identified, which expression was significantly associated with prognosis. Our aim was to analyze differences in gene expression profiles between hereditary (BRCA1mut linked) and sporadic breast cancer and try to find molecular basis underlying clinical differences in those diseases observed by some authors. We used HG U133 Plus 2.0 oligonucleotide microarrays allowing detection of 47 000 transcripts. Gene expression profiling was performed in following samples: i. - eight cases of medullary breast carcinoma, five with mutations in BRCA1 gene and three without mutations; ii - six cases of ductal carcinoma, three with mutated BRCA1 and three with wild type BRCA1; iii - four samples of normal breast tissue. Samples were normalized by RMA (Robust Microarray Analysis) method. At first step, we performed Principal Component Analysis (PCA), which confirmed that the distance of breast tumors to normal tissues is large enough to be detected by unsupervised method. We did not observe such a division in PCA results in respect to BRCA status and histopathology. Thus, supervised methods were applied. Among the genes that we found to be linked to BRCA status, three were previously reported by Hedenfalk et al. (N. Engl. J. Med. 2001, 344, 539-48), namely: selenium-dependent glutathione peroxidase 4 (GPX4), transducer of ERBB2 1 (TOB1; two transcripts related to BRCA status) and ARVCF (Armadllo repeat gene deleted in VCFS, catenin family). GPX4 and TOB1 were decreased in BRCA1-positive tumors, while ARVCF gene expression was slightly increased in this group. APPLICATIONS OF MIXTURE MODELS TO GENE EXPRESSION PROFILES. J. Polańska, M. Wiench, M. Jarząb, B. Jarząb, J. Rzeszowska-Wolny, M. Kimmel, A. Świerniak, A. Polański A very important topic in the research concerning DNA microarray data processing is modeling distribution of gene expression levels. One possible approach is using Gaussian mixtures for logarithms of expression levels. Approximating probability density functions (pdfs) of log expression levels by Gaussian mixtures can be used to solve several issues in interpretation of DNA microarray data. Decomposition of data into normal components is setting thresholds to classify expression levels as ''change'', ''no change'', ''overexpressed'', ''underexpressed'' etc., based on pdfs of estimated components. A related area is mixture modeling of values of sets of significance levels obtained from DNA microarray scanners and classical statistical analyses applied to gene expression profiles. It is formulated as the problem of estimating False Discovery Rate (FDR) to properly address the multiple testing phenomena in DNA microarrays. Recently a technique to solve the FDR problem by decomposing the distribution of significance levels to (at least) two components, beta and uniform and was described in the paper by Gadbury et al. (2004). The p-value distribution is modelled as a mixture of .+1distributions on the interval [0, 1]: 
where pi is the p-value from a test on the ith gene, p is the vector of k p-values, and .j is the probability that a randomly sampled p-value belongs to the jth component. If no genes are differentially expressed, the sampling distribution of p-values will follow a uniform distribution on the interval [0,1]. If some genes are differentially expressed, additional beta distribution components will be required to model a set of p-values clustering near 0. Maximum likelihood estimates of the parameters can be obtained using numerical methods. The decomposition applied to data on thyroid cancer is shown in the plots below. 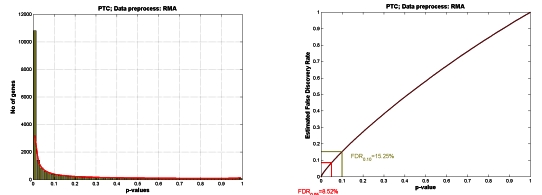
Figure in the left hand side shows the quality of the decomposition into beta and uniform components (red curve versus histogram). Using the decomposition the plot in the right can be computed which allows the following conclusion to be drawn: setting the threshold for a single test equal to 0.05 will result in 8.5% of FDR. Further analysis will lead to estimating TP and TN levels equal to TN=65% and TP=97%. Increasing the threshold to 0.1 will result in increasing FDR to 15.25% and TN to 75% and changing TP to 95%. Acknowledgement: This work was supported by grants no PBZ/KBN/040/P04/2001 and 4T11F01824. GENE EXPRESSION PROFILES AND CELLULAR STRESS Joanna Rzeszowska-Wolny1, Joanna Polańska2, Robert Herok1, Maria Konopacka1, Ronald Hancock3 1Department of Experimental and Clinical Radiobiology; 2Institute of Automation, Silesian University of Technology, 44-100 Gliwice, Poland; 3Laval University Cancer Research Centre, Quebec, Canada Using oligonucleotide microarrays we studied the changes of gene expression in response to ionizing radiation treatment of human erythroleukemic cells K562. In analyzing the results we used a new approach utilising Gaussian mixture model to describe total gene expression profiles obtained in all hybridizations and to compare expression in differentially treated cells. Metropolis-Hastings algorithm was used to determine the best fitting Gaussian components that describe experimentally obtained histograms. The Gaussian component patterns were characteristic for cell type and changed after irradiation with the dose of 4 Gy. The histograms describing frequencies of hybridization signal level in control K562 were decomposed into five fractions. Among genes found in low signal level fractions #1, #2 and #3 prevailed receptor genes, signal transduction proteins and transcription factors. Fractions #4 and #5 contained mainly genes for proteins taking part in translation process. Gaussian mixture model applied in two-sample comparisons allowed to identify genes that significantly changed their expression after irradiation. Ionizing radiation caused synchronous down-regulation of more than 2700 genes mainly from fraction #2. The mean transcription level of genes in this fraction is about 2 molecules per cell. The model of populations with non-uniform gene expression will be discussed. CD150-MEDIATED SIGNALING S. P. Sidorenko1, S. V. Mikhalap1, L. M. Shlapatska1, O. V. Yurchenko1, M. Y. Yurchenko1, G. G. Berdova1, K. E. Nichols3, E. A. Clark2 1Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology, NAS of Ukraine, Kiev, Ukraine, 2University of Washington, Department of Immunology, Seattle 98195, USA 3Children's Hospital of Philadelphia, Department of Pediatric Oncology, Philadelphia PA, USA The CD150 is a prototypic receptor of a subfamily of dual-function coreceptors within CD2 family. The CD150 subfamily consists of CD150 along with CD84, CD229, CD244, NTB-A/SF2000, and CS1/CRACC. The genes of CD150 subfamily are clustered together on a long arm of chromosome 1 at bands 1q21-24. The criteria for defining this subfamily are the presence of at least two characteristic signaling motifs TxYxxV/I (ITSM - immunoreceptor tyrosine-based switch motif) in the cytoplasic tail of the receptor and binding of adaptor proteins SH2D1A and/or EAT-2 to these ITSMs. The ITSM is different from immunoreceptor tyrosine-based motifs ITAM and ITIM. Like ITIM, ITSMs are tyrosine phosphorylated by Src-family kinases and serving as binding sites for SH2-containing phosphatases: SHP-1, SHP-2 and SHIP. Unlike ITIM, ITSMs bind directly adaptor proteins SH2D1A and EAT-2, Src-family kinases Fyn(T), Lyn, Fgr and the p85 subunit of PI3-kinase. The paired ITSM within cytoplasmic tails of the receptors contribute to the dual-function of CD150 subfamily receptors. At least two signal transduction pathways are initiated via CD150 subfamily receptors. Erk1/2 pathway requires Ras, Raf, MEK1/2 and SHIP, but not SH2D1A. Both SHP-2 and SHIP may contribute to CD150-mediated Erk-activation. CD150 ligation also results in PI3K-dependent Akt phosphorylation. This pathway depends on SH2D1A and Syk expression; however it is SHIP-independent and is negatively regulated by Lyn, Btk and SHP-2. Taken together, CD150 subfamily receptors mediate different functions depending on the availability of downstream molecules within their signal transduction pathways, and SH2D1A functions as a molecular switch. Analysis of CD150 and SH2D1A expression in non-Hodgkin's and Hodgkin's lymphomas revealed stages of B cell differentiation where these molecules are expressed alone or coexpressed. Signaling studies in Hodgkin disease cell lines showed that CD150 is linked to the ERK and Akt pathways in neoplastic B cells. Our data support the hypothesis that CD150 and SH2D1A are coexpressed during a narrow window of B cell maturation and SH2D1A may be involved in regulation of B cell differentiation via switching of CD150-mediated signaling pathways. Supported by INTAS grant 011-2883 (to S.P.S.), U.S. Civilian Research and Development Foundation Grants UB2-531 and UB2-2443-KV-02 (to S.P.S.) THE ROLE OF MOLECULAR CHAPERONS IN CANCER L. Sidorik1, R. Kyyamova1, L. Kapustian1, V. Filonenko1, P. Pogrebnoy2, D. Litvin2,T. Dudchenko2, V. Lyzogubov3, V. Usenko3 1Institute of Molecular Biology and Genetics NAS of Ukraine, Kiev, Ukraine 2Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology NAS of Ukraine, Kiev, Ukraine 3Morphological Laboratory "BIONTEK", Dnipropetrovsk, Ukraine Physiological stresses (heat, hemodynamics, genetic mutations, oxidative injury and myocardial ischemia) produce pathological states in which protein damage and misfolded protein structures are a common denominator. The specialized proteins family of antistress proteins - molecular chaperons (HSPs) - is responsible for correct protein folding, dissociating of protein aggregates and transport of newly synthesized polypeptides to the target organelles for final packaging, degradation or repair. They are inducible in different cellular processes such as division, apoptosis, signal transduction, differentiation and hormonal stimulation. HSPs are involved in numerous diseases including cancer, revealing changes of expression and cell localization. The investigation of molecular and cellular mechanisms of anticancer immune response has led to the discovery that some members of HSPs family induce specific protection against cancer. Recently it has been observed in rodent models that autologous tumor-derived HSP-peptide complexes were able to induce a specific anti-tumor immune response, i.e. were effectve in adjuvant setting. Moreover, the overexpression of some HSPs (as Hsp70, Hsp60 and Hsp27) in membranes of some tumors (such as breast cancer, endometrial-cervix cancer) correlated with poor prognosis and resistance to therapy while in osteosarcoma, squamous cell carcinoma of the esophagus and renal cell carcinoma the Hsp70 over-expression was associated with improved prognosis and response to chemotherapy. The specific anti-HSPs autoantibodies level correlated in some cases with the target organ injury degree. We studied changes of the main mitochondrial chaperon Hsp60 expression level and re-localization in cells of thyroid cancer as well as accompanying autoimmune processes using Western-blot and immunohistochemical analysis. Positive correlation between Hsp60 expression level, anti-Hsp60 autoantibodies level and thyroid gland injury degree has been observed. These changes correlated with re-localization of Hsp60 (anomal expression on the tumor cells surface) and (in many cases) with drug-resistance of patients investigated. Based on the presented and literature data we have proposed a new approach to the development of an effective therapeutic and diagnostic tool for cancer treatment. Corresponding author: Dr.Sidorik L.L., PhD Institute of Molecular Biology and Genetics NAS of Ukraine, 150 Zabolotnogo str, 03143, Kiev-143, UKRAINE Phone: 38 044 2665589 FAX: 38 044 2660759 E-mail: sidorik@imbg.org.ua SVD AS A TOOL FOR PATTERN DISCOVERY IN GENE EXPRESSION DATA. Krzysztof Simek Institue of Automatic Control, Silesian University of Technology, Akademicka 16, 44-100 Gliwice, Poland. Singular Value Decomposition (SVD) is a matrix factorization known from linear vector algebra, which reveals many important properties of a matrix. It is a standard tool in many areas of physical sciences, and many algorithms in matrix algebra make use of SVD. Recently, gene expression data were analyzed using Singular Value Decomposition. In gene expression data analysis the principal aim of application of SVD is to detect and extract internal structure existing in the data and corresponding to important relationships between expression of different genes. We aim to investigate the effectiveness of SVD as a preprocessing step to cluster analysis of gene expression data. In the clustering literature, SVD is sometimes applied to reduce dimensionality of the data set prior to clustering. The idea behind using SVD prior to cluster analysis is that SVD may extract the cluster structure in the data. Since characteristic modes are uncorrelated and ordered, the first few that are the most significant and reflecting most of data variation are usually used in cluster analysis. Our approach differs from that known from the literature, where characteristic mode coefficients (gene coefficient vectors), are used for clustering instead of original variables. We propose to apply SVD to select a set of original genes and then apply them for clustering samples by one of standard algorithms. The proposed gene selection algorithm inspects gene coefficient vectors corresponding to the set of the most significant characteristic modes. Each coefficient is compared to the threshold value, the meaning of which is similar to a 3 o statistical significance cutoff. If the magnitude of the element is greater than the threshold, the corresponding gene is selected to the clustering set. In practice we choose genes having sufficiently big coefficients for the most important characteristic modes. Variation of the threshold value gives possibility of changing a number of selected genes. In the result we obtain a set of genes having patterns 'similar' to the dominant modes. We test our approach on three different sets of biological gene expression data aquired in in the Comprehensive Cancer Centre Maria Skłodowska-Curie Memorial Institute Branch Gliwice, Poland, using Affymetrix Human Genome U133A arrays. This work was partially supported by grant BW-424/RAu1/04. FAST DENOISING OF cDNA MICROARRAY IMAGES Bogdan Smołka Silesian University of Technology, Akademicka 16, 44-100, Gliwice, Poland The cDNA microarray is an effective tool for simultaneously assaying the expression of large numbers of genes and is perfectly suited for the comparison of gene expressions in different populations of cells. A cDNA microarray is a collection of spots containing DNA, deposited on the surface of a glass slide. The spots occupy a small fraction of the image area and they have to be individually located and isolated from the image background prior to the estimation of its mean intensity. The fluorescent intensities for each of the spots are measured using two light sources of different wavelength, producing a two-channel image. The image is false colored using the red and green color for each image components, which represent the light intensity emitted by the two fluorescent dyes used in this method. The quantitative evaluation of microarray images is a difficult task. The major sources of uncertainty in spot finding and measuring the gene expression are variable spot sizes and positions, variation of the image background and various image artifacts. Additionally the natural fluorescence of the glass slide and non-specifically bounded DNA or dye molecules add a substantial noise floor to the microarray image. To make the task even more challenging, the microarrays are also afflicted with discrete image artifacts such as highly fluorescent dust particles, unattached dye, salt deposits from evaporated solvents, fibers and air-borne debris. So, the task of microarray image enhancement is of paramount importance. In this work a novel, ultrafast noise reduction method for the enhancement of multichannel images is presented. This novel technique can be successfully applied for the denoising of the cDNA microarray images, as those pictures consist of two channels and are of huge dimensions, which makes their processing very time consuming. The proposed method is a simplification of the so called Peer Group Filtering. Instead of calculating the adaptive threshold, which determines the number of pixels similar to the central pixel in the filtering window, a global threshold for the similarity function can be experimentally determined. Additionally, for not too high noise intensities, the number of pixels belonging to the peer group can be established. In this way the algorithm calculates the number of pixels in the filtering window, which satisfy a certain similarity criterion and checks if this number is below a certain, experimentally established threshold value. If this is the case, then the central pixel is being replaced by the local vector median of the samples, otherwise the central sample is retained unchanged. The new technique is capable of reducing various kinds of noise present in the microarray images and it enables efficient spot localization and estimation of the gene expression level due to the smoothing effect and preservation of the spot edges. The presentation will include comparison of the new technique of noise reduction with the standard procedures used for the processing of vector valued images, as well as examples of the efficiency of the new algorithm when applied to typical cDNA microarray images. PROCESSING OF LIPID PEROXIDATION-DERIVED DNA ADDUCTS BY REPAIR ENZYMES AND DNA POLYMERASES Barbara Tudek, Beata Rusin, Leena Maddukuri, Marek Komisarski, Jarosław T. Kuśmierek, Paweł Kowalczyk, Jarosław Cieśla Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, 02-106 Warsaw, Poland; e-mail: tudek@ibb.waw.pl Lipid peroxidation leads to the formation of a large family of alkenals, among which trans-4-hydroxy-2-nonenal (HNE) is one of the most ubiquitous. HNE forms adducts to all four DNA bases, however with different efficiency: G>C>A>T. These are cyclic propano- or ethenoadducts bearing a hexyl- or heptyl- side chains. Due to the fact that reaction of aldehyde group with the nitrogen atom of DNA base is reversible, HNE can also form inter- strand DNA cross-links, and these constitute around 5% of damaged DNA. Interaction of HNE with DNA is sequence-specific. In vitro studies on p53 gene sequence show that the major hot-spots for p53 mutations are readily modified by HNE, with the possibility of cross-links formation within three hot-spot codons 175, 248 and 249. HNE-induced modifications block DNA synthesis, and reveal mutagenic properties in bacterial and mammalian cells. In bacteria, the major target for mutations are HNE adducted cytosine residues. HNE-DNA lesions when present in single-stranded DNA, are highly recombinogenic. Removal of these lesions from the double-stranded DNA is realized mainly by the nucleotide excision repair pathway. Equally important mechanism of avoiding deleterious effects of HNE-adducts presence in DNA is translesion synthesis by damage-specific DNA polymerases. E.coli DNA polymerase IV is engaged in lesions bypass, probably in an error-free manner. DNA polymerase V is indispensable for induction of mutations. Human cells deficient in nucleotide excision repair, both global (XPA) and transcription-coupled (CSB) are more sensitive, and develop higher level of sister chromatid exchanges (SCE) than their repair proficient counterparts under low, micromolar HNE concentrations. Similar are Chinese hamster cells deficient in nucleotide excision repair (XPD) and non-homologous end joining (NHEJ). Interestingly, transcription-coupled repair deficient line (CSB) appear to be more sensitive (10-100 fold) to HNE than any other tested cell line. Thus NER and NHEJ pathways are engaged in processing of the HNE-induced DNA damages in mammalian cells. HNE-induced DNA damages, if not repaired, may block replication, transcription and trigger recombination in mammalian cells. Hsp90 AND MDM2 ARE IMPORTANT FOR THE TRANSCRIPTIONAL ACTIVITY OF WILD-TYPE p53 Bartosz Wawrzynow, Dawid Walerych, Grzegorz Kudla, Malgorzata Gutkowska, Aleksandra Helwak, Joanna Boros, Alicja Zylicz and Maciej Zylicz International Institute of Molecular and Cell Biology in Warsaw, Institute of Experimental Biology PAS, Institute of Biochemistry and Biophysics PAS The p53 tumour suppressor protein is a transcription factor which regulates cellular response to stress, abnormal cell proliferation and DNA damage. In a non-stress situation, the level of p53 in the cell is low, mainly regulated at the posttranslational level by the E3 ubiquitin ligase, MDM2. Hsp90 is known to interact with both p53 and MDM2 but the role of this interaction is not clear. Immortalized human fibroblasts were used to investigate the interactions of the Hsp90 molecular chaperone with the wild-type p53 tumor suppressor protein. We show that geldanamycin or radicicol, specific inhibitors of Hsp90, diminish specific wild-type p53 binding to the p21 promoter sequence. Consequently, these inhibitors decrease p21 mRNA levels. This leads to a reduction in cellular p21/Waf1 protein, known to induce cell cycle arrest. To support our in vivo findings, we used a reconstituted system with highly purified recombinant proteins to examine the effects of Hsp90 on wild-type p53 binding to the p21 promoter sequence. We show that the incubation of recombinant p53 at 37oC decreases the level of its wild-type conformation and strongly inhibits the in vitro binding of p53 to the p21 promoter sequence. Interestingly, hHsp90 alfa in an ATP-dependent manner can positively modulate p53 DNA binding after incubation at physiological temperature of 37oC. Consistent with our in vivo results, geldanamycin can suppress Hsp90 ability to regulate in vitro p53 DNA binding to the promoter sequence. We have shown that Hsp90 partially unfolds p53. Unfoldase activity of Hsp90 requires the presence of ATP. Surprisingly, MDM2 accelerates this reaction. Moreover, MDM2 alone possesses the chaperone activity. We propose that partial unfolding of p53 catalysed by Hsp90 and MDM2, and the subsequent spontaneous refolding of p53 back into a wild-type-like conformation, may prevent p53 aggregation thus increasing the p53-DNA binding. These events would ultimately promote the p53 transcriptional activity and allow for the ubiquitination and degradation of p53 protein. THE OTHER FACE OF HISTONE H1 Piotr Widłak, Magdalena Kalinowska, Missag H. Parseghian, Xu Lu, Jeffrey C. Hansen and William T. Garrard Histone H1 is a multigene protein family, consisting of six different proteins in somatic cells of mammals. Histone H1 is an essential protein involved in regulation of chromatin structure and gene expression. Most recently a specific histone H1 subtype was demonstrated to be a messenger from the nucleus to the cytoplasm, signaling DNA damage and triggering apoptosis. Upon DNA damage, histone H1.2 was shown to leak to the cytoplasm, trigger the release of cytochrome c from mitochondria and initiate apoptosis. The apoptotic nuclease, DNA fragmentation factor (DFF40/CAD), is primarily responsible for internucleosomal DNA cleavage during the terminal stages of programmed cell death. Previously we demonstrated that histone H1 greatly stimulates naked DNA cleavage by this nuclease. In this work we investigate the mechanism of this stimulation with purified, recombinant, and synthetic mouse and human histone H1 species. We find that each of the six somatic cell histone H1 subtypes, which differ in primary sequence, equally activate DFF40/CAD. Using a series of truncation mutants of recombinant or synthetic mouse histone H1-0 species, we demonstrate that nearly as efficient stimulation can occur with different ~50- to 72-amino acid long fragments of the C-terminal domain (CTD). We show further that histone H1 C-terminal segments bind to DFF40/CAD and confer upon it an increased ability to bind to DNA. We conclude that the interactions identified here between histone H1 segments and DFF40/CAD effectively target linker DNA cleavage during the terminal stages of apoptosis. RET PROTOONCOGENE AND GENE EXPRESSION PROFILE OF THYROID CANCER Małgorzata Wiench Structural rearrangements of the RET gene leading to its oncogenic activation cause the neoplastic transformation in about 30% of papillary thyroid cancers (PTC). They are linked to radiation history on the one hand and to the young age of presentation on the other. It remains to be determined whether the same signal transduction pathways are stimulated in both RET-positive and RET-negative tumors, whether they occur on different levels of the same pathway (MAP kinase cascade), or rather there are quite different mechanisms underlying the follicular cell transformation. The purpose of this study was to evaluate whether RET-positive and RET-negative tumors show distinct patterns of gene expression. For gene expression studies the cRNA obtained from 12 RET-positive and 17 RET-negative PTC tumors were hybridized to Human Genome U133A arrays according to Affymetrix recommendations. The presence of RET rearrangement was confirmed by RT-PCR study. All data were obtained using MAS 5.0 software (Affymetrix). Comparison of gene expression between groups was performed by SAM (Significance Analysis of Microarrays). For further analysis, the list of 343 genes differentiating RET-positive and RET-negative tissues was chosen [5,02% False Discovery Rate (FDR) implies 17 falsely positive genes]. Among them there were 71 genes up-regulated in RET-positive and 272 genes up-regulated in RET-negative tissues. The Gene Ontology analysis revealed the striking over-representation of the cell communication-related and morphogenesis-related genes in the RET-positive cancers. In the group of RET-positive up-regulated genes there were 21 transcripts with at least two-fold change whereas only two transcripts (TPD52L1, RRAGD) fulfilled this criterion in the RET-negative over-expressed genes. A special attention was paid to signal transduction genes frequently represented in RET-positive PTCs: NFKBIA, RGS2, SLC2A3, GEM, NR4A2, NR4A3, OPCML, PASK as well as RET itself. The putative difference between RET-positive and RET-negative papillary thyroid cancers is not distinct enough to be deciphered by an unsupervised method (i.e. SVD). Nevertheless, the supervised selection is able to identify a panel of differentially expressed genes, mainly contributing to cell communication and signal transduction processes in tumors initiated by RET rearrangement. DO INTERINDIVIDUAL DIFFERENCES IN LYMPHOCYTE CHROMOSOME RADIOSENSITIVITY EXIST? Andrzej Wójcik Institute of Nuclear Chemistry and Technology, Department of Radiation Biology and Health Protection, Warszawa, and Swietokrzyska Academy, Institute of Biology, Department of Radiobiology and Immunology, Kielce, Poland. Individuals show marked differences in radiation sensitivity. This has considerable consequences both in the fields of radiation protection and radiation therapy. Despite numerous studies no single, reliable assay has been established that reflects the intrinsic, individual sensitivity. Human peripheral blood lymphocytes make up the biological system of choice. The endpoints used most frequently are chromosomal aberrations, micronuclei or kinetics of DNA repair. Each assay has its advantages and pitfalls. We are presently studying the frequencies of radiation-induced aberrations in lymphocytes of patients with larynx carcinoma. Furthermore we search for a possible correlation between the kinetics of DNA repair measured by the alkaline comet assay and the frequency of chromosomal aberrations. Finally, we study, by chromosome painting, the extent of interindividual differences in the sensitivity of selected chromosomes in lymphocytes of healthy donors. Results will be presented and discussed with reference to published data. | |
| Ostatnia aktualizacja strony |