| This is an old, no longer maintained version of the website, for new one please visit: http://gsn.io.gliwice.pl |

 |

Gliwice Scientific Meetings 2010 ~~~ Lecture abstracts ~~~
Session I:
SIMULATED SUNLIGHT-INDUCED DAMAGE TO MITOCHONDRIA AND mtDNA IN HUMAN SKIN CELLS Luciene Maria Zanchetta1, F. Lyng2, J. Walsh3, J.E.J. Murphy1
1Mitochondrial Biology & Radiation Research Group, Institute of Technology Sligo, Ballinode, Sligo, Ireland; 2Radiation and Environmental Science Centre, Focas Institute, Dublin Institute of Technology, Kevin St, Dublin 8, Ireland; 3School of Physics, Dublin Institute of Technology, Kevin St., Dublin, Ireland, lucienemz@gmail.com.
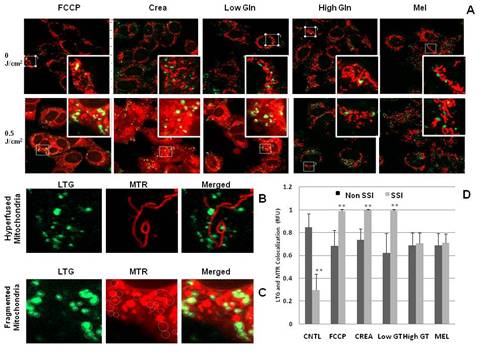
Solar Radiation (SR) causes cell damage or death by disrupting cellular energy metabolism at multiple levels directly altering DNA and protein structures and by increasing production of reactive oxygen species (ROS). SR can induce damage in mitochondrial DNA (mtDNA) that may lead to mitochondrial dysfunction which has been associated with skin cancer progression. MtDNA is organized in nucleoids containing several copies of the genome. Changes in mitochondrial dynamics, more precisely mitochondrial fusion disruption, are associated with mtDNA nucleoid loss and decreased mitochondrial respiratory function. Damaged mitochondria are recycled thought mitophagy. Metabolic energy sources have been linked to mitochondrial dynamics (fusion, fission and biogenesis ratio) changes. The objective of this study was to assess simulated sunlight-induced changes in cellular, mitochondrial and mtDNA end-points to further investigate the role of these essential organelles in the response to the main environmental stressor associated with inducing skin cancer. A Q-Sun Solar Simulator (Q-Lab, USA) was employed to expose cultured human skin cells to Simulated Sunlight Irradiation (SSI). Cells were also bio-energetically challenged and their mitochondrial activity inhibited or stimulated. Cellular analyses were: viability; apoptosis; Reactive Oxygen Species (ROS) and total DNA and protein content. Mitochondrial analyses included: mitochondrial membrane potential (MMP); mass and morphology; recycling by autophagy; mtDNA damage and repair using conventional, Long Range and Real Time PCR; mtDNA nucleoid number and mitochondrial localization and dynamics. Decrease in MMP, ROS, mitochondrial mass, mtDNA levels and changes in nucleoid number and distribution were observed in human skin cells post SSI, suggesting that mitochondrial dynamics may play an important role in cellular responses to solar radiation damage. The common deletion mtDNA4977, though detected, did not directly increase in frequency with sunlight exposure though the mtDNA3895 deletion, previously found to be associated with sunlight exposure, was observed to be substantially increased in a cell-type and dose-dependent manner in skins cells post SSI. Increases in mitochondrial genome number and mtDNA3895 were observed as an early response to low-dose SSI in human skin cells. Changes in relative mitochondrial mass did not correlate with relative mitochondrial genome number per cell. Glutamine and melanin were observed to reduce mitophagy and to prevent increases in mtDNA deletions ratio post SSI likely via mitochondrial fusion/hyperfusion stimulation and/or mitochondrial genome protective action. Impaired mitochondrial bioenergetics, dynamics and recycling may play a significant role in the melanoma tumour initiation and progression in humans post systematic sunlight over-exposure. Furthermore the sensitive nature of the mitochondrial population of skin cells should not be underestimated as dynamic changes in their biology are evident even in cell populations that received low level irradiation of simulated sunlight.
LOW DOSE RADIATION INDUCED TRANSCRIPTIONAL ALTERATIONS IN BYSTANDER PRIMARY HUMAN FIBROBLAST CELLS
G. Sáfrány, H. Hegyesi, N. Sándor, B. Schilling, K. Lumniczky
Frédéric Joliot-Curie National Research Institute for Radiobiology and Radiohygiene, Budapest, Hungary.
Introduction: Formerly, we studied transcriptional alterations in primary human fibroblast cells after irradiation with 2 Gy (Kis et al. Int J Radiat Oncol Biol Phys. 66:1506-14. 2006). Thirty consensus radiation response genes answered to radiation in identical manner in all investigated cells. Now, we have investigated low dose radiation induced transcriptional responses in directly hit and bystander cells. Based on the studies two genes (GDF15, TP53INP1) were selected to investigate the effect of gene silencing in bystander response. Methods: F11 primary human fibroblasts were irradiated with different doses (10, 40, 100 and 500 mGy) of 60Co gamma radiations. To investigate radiation-induced transcriptional alterations in directly irradiated cells, RNA was isolated 2 h after irradiation. To study responses in bystander cells the culture medium was removed from the irradiated cells 2 h after irradiation and transferred to unirradiated recipient cells. The transcriptional profile was analyzed by whole genome microarrays. Time and dose dependent alterations were validated by quantitative RT-PCR. For gene silencing the MISSION Lentiviral Transduction system (5 different shRNA constructs) was applied. Results: When cells were irradiated with 500 mGy 1119 genes responded to radiation. Ten of the formerly identified consensus radiation response genes changed its transcription (CDKN1A, TP53INP1, CYP26B1, BTG2, BBC3, PPM1D, THSD1, GDF15, NM_024661, BC010544). Irradiation of F11 fibroblasts with 100 and 10 mGy altered the transcription profile of 847 and 1414 genes, respectively. When we compared the transcription profile of cells irradiated with 500 and 100 mGy 377 similar alterations were detected, among them 6 consensus radiation response genes (CDKN1A, TP53INP1, GDF15, BTG2, BBC3, NM_024661). In bystander cells 655 and 406 genes responded to 500 and 100 mGy irradiations on the transcription level. After irradiation with 40 and 10 mGy the number of responding genes were 152 and 619. When we compared the responses in bystander cells after irradiation with 100 and 40 mGy only 40 genes responded identically. The comparison of the transcriptional profile of 40 and 10 mGy irradiated cells detected 60 similar responses. In directly irradiated fibroblast cells GDF15 expression increased with the applied dose. Transcription reached the highest level 2 hours after irradiation, then decreased with time, although increased expression was still detectable 48 hours after irradiation. In bystander cells GDF15 did not alter its transcription two hours after the addition of conditioned medium. Interestingly, at later time points (24 and 48 hours) we detected decreased transcription levels. With one shRNA construct we could suppress GDF15 expression in fibroblast cells to 25-30% of the wild type level. Radiation response of the GDF15 gene was suppressed in GDF15 silenced cells. Radiation sensitivity of the GDF15 silenced cells increased by about 1.5-fold. The bystander response disappeared in GDF15 silenced cells. Conclusions: By the analysis of radiation induced transcriptional alterations one might find potential biomarkers suitable to detect low dose responses. We could only partially silence the GDF15 gene using shRNA constructs. Still, the results suggest that GDF15 gene silencing affects the radiation sensitivity of the cells; it influences bystander effects and alters radiation-induced gene expression profiles. ISSUES IN THE INTERPRETATION OF LOW DOSE EFFECTS IN RADIOBIOLOGY AND ENVIRONMENTAL RADIATION PROTECTION
Carmel Mothersill, Colin Seymour
Medical Physics and Applied radiation Sciences Department, McMaster University, Hamilton, Ontario, Canada L8S 4K1, email: mothers@mcmaster.ca.
The last 15 years have seen a major paradigm shift in radiation biology. Several discoveries challenge the DNA centric view which holds that DNA damage is the critical effect of radiation irrespective of dose. This theory leads to the assumption that dose and effect are simply linked – the more energy deposition, the more DNA damage and the greater the biological effect. This is embodied in radiation protection (RP) regulations as the linear-non-threshold or LNT model. However the science underlying the LNT model is being challenged particularly in relation to the environment because it is now clear that at low doses of concern in RP, cells, tissues and organisms respond to radiation by inducing responses which are not predictable by dose. These include adaptive responses, bystander effects, genomic instability and low dose hypersensitivity/induced radioresistance and are commonly described as stress responses, while recognizing that “stress” can be good as well as bad. The phenomena contribute to observed radiation responses and appear to be influenced by genetic, epigenetic and environmental factors, meaning that dose and response are not simply related. The big question is whether our discovery of these phenomena means that we need to re-evaluate RP approaches. This is the subject of presentation. On the one hand the mechanisms mean that low dose radiobiology is very complex and supra linear or hormetic responses are equally probable but their occurrence is unpredictable for a given individual. On the other hand, the bottom line is that epidemiology does not suggest big effects in either direction at low doses. Issues which may need consideration are synergistic or antagonistic effects of other pollutants because RP at present only looks at radiation dose but the new radiobiology means that chemical or physical pollutants which interfere with tissue responses to low doses of radiation could critically modulate the predicted risk. Similarly, the “health” of the organism could determine the effect of a given low dose by enabling or disabling a critical response. These issues will be discussed. POTENT BYSTANDER EFFECTS INDUCED BY TARGETED RADIONUCLIDES
Rob Mairs
Cancer Research UK Beatson Laboratories, Garscube Estate, Glasgow, Scotland.
Radiation as a cancer modality is of high physical precision but limited biological specificity. Targeted radiotherapy, the delivery of radiation to cancer cells by radionuclides conjugated to tumour-seeking agents, is a biologically attractive option. The radiopharmaceutical [131I]meta-iodobenzylguanidine ([131I]MIBG) is an effective single agent for the treatment of neuroblastoma. However, uptake of the drug in malignant sites is insufficient to cure disease. Non-uniform distribution of radiopharmaceuticals in tumours is a major constraint upon the efficacy of targeted radionuclide therapy. It may be possible to compensate for heterogeneity of uptake of [131I]MIBG, resulting in underdosing of some tumour regions, by exploiting radiation-induced biological bystander effects deriving from the cellular processing of the physical radiation insult. This phenomenon may play an important part in the overall efficacy of radionuclide targeting. We examined this effect using media transfer methodology. Medium from cells that accumulated radiopharmaceutical was transferred to cells which had not been exposed to radioactivity and clonogenic survival was determined in donor and recipient cultures. We observed that potent toxins were generated specifically by cells which concentrated radiohalogenated MIBG. These were LET-dependent and distinct from those elicited by conventional radiotherapy. Recently we have been characterising the nature of radionuclide-induced bystander signals and determining the dependence upon genotype (e.g. P53 status) of the efficiency of this mode of kill in tumour cells. Elucidation of the pathways involved in the generation of factors by radionuclide-concentrating tumour cells could indicate ways of manipulating bystander signal production to reduce toxicity to normal tissues that may be inadvertently irradiated during the course of a targeted radiotherapy regime. With the growing appreciation of the significance of radiation-induced bystander effects, it is becoming clear that these must be rigorously studied with respect to their chemical nature, tumour specificity, and dose- and time-dependence.
OXIDATIVE STRESS AND BYSTANDER EFFECT
Joanna
Rzeszowska-Wolny1,2, Artur Cieślar-Pobuda1, Maria Wideł1,
Roman Jaksik1,
1Systems Engineering Group, Silesian University of Technology, 44-100
Gliwice;
Cells exposed to ionizing radiation release factors which induce DNA damage, chromosomal instability, apoptosis and changes of transcript levels in neighboring untreated cells, the phenomena known as bystander effects. The intercellular signals that cause bystander effects are as yet poorly defined, and a number of studies suggest that up-regulation of oxidative metabolism in non-targeted cells is involved. We examined damage in nucleic acids, changes of transcript profiles, and levels of reactive oxygen species in a few human cell lines at different time points after direct irradiation, or in bystander cells growing in culture medium containing factors released by irradiated cells. A short time after treatment both increase and decrease of transcript levels were observed, and the number of up and down-regulated transcripts and functional pathways to which they belonged was very similar in both irradiated and bystander cells. At the same time, an increase in 8-oxo-7,8-dihydro-guanosine (8-oxoG) in RNA and changes in the level of reactive oxygen species (ROS) were observed in the cells. Because binding of miRNAs and proteins to mRNAs are important factors in regulating mRNA stability, we explored if the up- or down-regulation of transcripts is correlated with the presence of sequence motifs which bind miRNAs and proteins. In all the cell lines exanined more transcripts were up- than down-regulated 1 h after irradiation. The up-regulated transcripts contained significantly more (p<10-10) target motifs for miRNAs and also, in three cell lines, for protein-binding AU-rich motifs in their 3' untranslated regions compared with those down-regulated or unchanged. These results are consistent with the model that an increase in ROS induced by irradiation or by signals released from irradiated cells can cause oxidative damage to RNAs, which modulates their specific interactions with miRNAs or mRNA- binding proteins and thus causes changes in mRNA stability.
THE ONCOGENIC POTENTIAL OF ADAPTOR PROTEIN Ruk/CIN85 IN HUMAN BREAST ADENOCARCINOMA
L.B. Drobot
Palladin Institute of Biochemistry, National Academy of Sciences of Ukraine, Kiev, Ukraine.
Adaptors are proteins of multi-modular structure without enzymatic activity. Their capacity to organize large, temporary protein complexes by linking proteins together in a regulated and selective fashion makes them of outstanding importance in the establishment and maintenance of specificity and efficiency in all known signal transduction pathways. Given the important role of adaptor proteins in propagating cellular signals, it is quite likely that their dysfunction may be involved in carcinogenesis. The adaptor/scaffold protein Ruk/CIN85, containing multiple SH3 domains, was implicated in carcinogenesis by influencing a number of processes such as cell adhesion, motility and apoptosis. Although Ruk/CIN85 appears to modulate tyrosine kinase receptors and PI3 kinase signalling, the exact molecular mechanisms by which Ruk/CIN85 affects carcinogenesis are largely unknown. Using Western-blot analysis, a statistically significant increase in the expression level of Ruk/CIN85 full-length form was detected in human invasive ductal breast adenocarcinoma samples in comparison with surrounding conditionally normal tissues. Therefore, we decided to investigate the oncogenic potential of Ruk/CIN85 by overexpressing the full-length isoform in weakly invasive MCF-7 breast adenocarcinoma cells. The Rukl/CIN85 overexpressing cells showed a slower growth rate, decreased cell adhesion, and an enhanced anchorage-independent growth in soft agar. Furthermore, overexpression of Rukl/CIN85 also affected EGF-dependent signalling: activation of both Akt and ERK1/2 was faster than in the control cells and both kinases remained in their active state for up to 30 min after EGF treatment. Transwell migration and wound healing assays revealed that Rukl/CIN85 overexpressing cells possessed increased motility. The EGF-induced motility was attenuated in Rukl/CIN85-overexpressing cells but could be restored upon knock-down of Rukl/CIN85 with specific shRNA. It was found also that Ruk/CIN85 induced PAI-1 mRNA and protein expression both under normoxia and hypoxia. The induction of PAI-1 expression by Ruk/CIN85 occurred at the transcriptional level since the half-life of PAI-1 mRNA was not affected in cells overexpressing Ruk/CIN85 and reporter gene assays using wild-type and mutant human PAI-1 promoter luciferase constructs showed that the hypoxia responsive element was responsible for Ruk/CIN85 effects. Further, knocking down HIF-1α abolished not only the hypoxia-dependent but also the Ruk/CIN85-dependent PAI-1 induction. In addition, transient or stable overexpression of Ruk/CIN85 also induced HIF-1α protein levels and HIF-1 activity and knocking down Ruk/CIN85 reversed these effects. Thereby, Ruk/CIN85 interfered with the proline hydroxylation-dependent HIF-1α protein destabilisation. Together, these findings suggest that high levels of Rukl/CIN85 can modulate EGF-and hypoxia-dependent signalling and contribute to the conversion of breast adenocarcinoma cells into a more malignant phenotype.
Session
II: TRANSCRIPTION BY RNA POLYMERASE I AS TARGET FOR ANTICANCER THERAPY
Ross Hannan
Research Division, Peter MacCallum Cancer Centre, Melbourne, Australia.
Increased transcription of the ribosomal genes (rDNA) by RNA Polymerase I (Pol I) is a common feature of human cancer and enlarged nucleoli, indicative of accelerated rDNA transcription rate, have long been closely associated with transformation and tumour aggressiveness. Despite these observations no studies have directly examined the requirement for dysregulated rDNA transcription in the maintenance of the malignant phenotype. Here we show that increased rDNA transcription is necessary for MYC oncogenic activity and can be therapeutically targeted to treat tumours in a genetic model of lymphomagenesis. We demonstrate that restoration of hyperactivated rDNA transcription rates in Eu-MYC/+ lymphoma cells to the levels observed in normal B cells does not lead to slow tumor cell growth as might be predicted, but instead results in a rapid induction of programmed cell death. The apoptotic response is not an indirect consequence of ribosome insufficiency but rather due to induction of the ribosome biogenesis surveillance pathway characterized by rapid nucleolar disruption and the subsequent activation of p53-dependent apoptotic signaling. Using a specific small molecule inhibitor of Pol I transcription (CX-5461) currently in pre-clinical development we show that malignant B cells have a heightened dependence on elevated rDNA transcription that can be exploited in vivo as a therapeutic target for treatment of lymphoma. Strikingly, CX-5461 therapy of mice transplanted with Eu-MYC/+ lymphomas, induces a period of complete disease remission while maintaining a normal B-cell population. Our work reveals a previously unproven paradigm that links hyperactivated rDNA transcription and nucleolar integrity to maintenance of aggressive tumours independent of ribosome levels. Critically, these results also demonstrate how activation of a ribosome biogenesis surveillance pathway by selective inhibition of rDNA transcription rate, can be used as a novel therapeutic target for the treatment of cancer. SYNTHESIS AND BIOLOGICAL ACTIVITY OF GENISTEIN GLYCOSIDES AND GLYCOCONJUGATES
G. Grynkiewicz1, W. Szeja2, J. Puchałka2, G. Węgrzyn3, A. Rusin4, Z. Krawczyk4
1Pharmaceutical Research Institute, Warsaw; 2Department
of Organic, Bioorganic
Throughout millenia soy remained one of the most important agricultural crops, utilized for food by majority of Asian populations. Today, soybeans are a global product and soy GMO cultivars are selected for quality of oil and protein, with less attention to non-nutrient constituents, such as saponins and isoflavone conjugates, both of similar physicochemical characteristic derived from their glycosidic structure. However, according to numerous epidemiological studies, these minor constituents may exert both significant and advantageous influence on human health [1]. Genistein, the main isoflavone aglycone among phenylpropanoid secondary metabolites present in soy, belongs to the group of the most intensively studied natural products, for which numerous molecular targets and various mechanisms of biological activity have been identified [2-3]. Although poorly bioavailable and characterized by sub-optimal physicochemical properties, this compound is still considered a drug candidate, currently being tested in numerous clinical trials, while its glycoside – genistin, occuring in all natural sources of the isoflavone – attracts incomparably less attention. In keeping with our general field of interest (comparison between glycosides and aglycones in various biological activity tests), natural and synthetic conjugates of genistein have been studied, particularly with respect to inhibition of glycosaminoglycan storage in the central nervous system, as well as to cytotoxicity against selected types of cells. The results revealed considerable potential of synthetic genistein glycosides and glycoconjugates and revived interest in the availability of various complex phenolic glycoconjugates designed according to medicinal chemistry guidelines [4-5]. Short review of synthetic methods focusing on application of unsaturated pyranose synthons will be given [6,7], and biological activity of unsaturated genistein glycosides and glycoconjugates will be discussed.
References: [1]. Setchell K.D., Cassidy A., Dietary isoflavones: biological effects and relevance to human health, J. Nutr., 1999, 129:758-767. [2]. Dixon R.A., Ferreira D., Genistein, Phytochemistry, 2002, 60:205-211. [3]. Dixon
R.A., Sumner L.W., Legume natural products: understanding and manipulating
complex pathways [4]. Piotrowska E., Jakobkiewicz-Banecka J., Tylki-Szymanska A., Wegrzyn G., Genistin-rich isoflavone extract in substrate reduction therapy for Sanfilippo syndrome, Curr. Ther. Res. Clin. Exp., 2008, 69:166-179. [5]. Rusin A., Gogler A., Glowala-Kosinska M., Bochenek D., Gruca A., Grynkiewicz G., Zawisza J., Szeja W., Krawczyk Z, Unsaturated genistein disaccharide glycoside as a novel agent affecting microtubules, Bioorg. Med. Chem.Lett., 2009, 19:4939-4943. [6]. Priebe W., Fokt I., Grynkiewicz G., Glycal derivatives, In: Glycosciences (Fraser-Reid B, Tatsuta T, Thiem J; eds.), Springer-Verlag, Berlin 2008, 699-736. [7]. Ferrier R.J., Zubkov O.A., Transformation of glycals into 2,3-unsaturated glycosyl derivatives, Organic Reactions, 62:571-736 (2003).
SYNTHESIS OF COMPLEX DERIVATIVES OF URIDINE AS POTENTIAL INHIBITORS OF GLYCOSYLTRANSFERASES
I. Wandzik1, T. Bieg1, M. Czaplicka1, G. Grynkiewicz2, E. Król3, J. Paszkowska1, W. Szeja1, B. Szewczyk1
1Silesian University of Technology,
Department of Organic Chemistry, Bioorganic Chemistry
Glycosyltransferases (GTs) are enzymes involved in the biosynthesis of oligosaccharides, polysaccharides and glycoconjugates [1]. Modulation of GTs activities by efficient inhibitors is promising for the control of various molecular recognition processes including bacterial or viral infection and tumor progression. Therefore selective inhibitors of GTs are of interest because they may lead to the development of novel therapeutic agents. Different approaches based on analogies with donor substrates, acceptor substrates and transition state, respectively, have been used to design potent inhibitors of GTs [2]. Among them the development of donor substrate analogues has received considerable attention. Although many compounds have been designed and synthesized, only few of them exhibited significant activity against GTs. Recently we have undertaken a study on the design and synthesis of potent inhibitors of GTs as analogues of donor substrates. The proposed structures were composed of 2-deoxy sugar units and uridine. The choice of structures for the synthesis was preceded by docking simulation studies [3]. The majority of GTs utilise donors containing uridine pyrophosphate leaving group (UDP). N-acetylglucosaminyltransferase I (GnT I) and beta-1,4-galactosyltransferase I (b4GalT I) are typical examples. Therefore we carried out docking of proposed structures to the active sites of both enzymes. Structures with the best affinity were synthesized. In order to construct target compounds, orthogonally protected glycal substrates and uridine were used [4, 5]. The stereoselective synthesis of these compounds was accomplished using the Falck-Mioskowski protocol [6]. All of the synthesized compounds were then tested as potential inhibitors in a competition assay against bovine milk b4GalT I using fluorescent acceptor b-GlcNAc-O-(CH2)6-dansyl as a substrate. None of the compounds displayed significant inhibitory activity at concentrations up to 2.4 mM. Fortunately, in an independently carried-out biological assays two of the synthesized compounds exhibited antiviral activity against classical swine fever virus; this can be associated with inhibition of glycosylation at the stage of glycan modification which is characteristic for mammalian cells [7].
References: [1]. A. Varki, R. D. Cummings, J. D. Esco, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler (Eds): Essentials of Glycobiology Second Edition, CSHL Press, 2009. [2]. K-H. Jung and R. R. Schmidt in: Carbohydrate-Based Drug Discovery, C.-H. Wong Ed., Wiley-VCh Verlag: Weinheim, 2003, Vol. 2, p.609. [3]. I. Wandzik, Acta Pol. Pharm., 65, 735 (2008). [4]. I. Wandzik and T. Bieg, Bioorg. Chem., 35, 401 (2007). [5]. I. Wandzik, T. Bieg and M. Czaplicka, Bioorg. Chem., 37, 211 (2009). [6]. V. Bolitt, Ch. Mioskowski, S.-G. Lee and J. R. Falck, J. Org. Chem., 55, 5812 (1990). [7]. E. Król, I. Wandzik, W. Szeja, G. Grynkiewicz, B. Szewczyk, Antiviral Res., 86, 154 (2010). Biological activities of synthetic genistein derivatives
Aleksandra Rusin1, Zdzisław Krawczyk1,2
1Centre of Translational Research Maria
Skłodowska-Curie Memorial Center and Institute
Genistein, the main isoflavone of soybean, attracts much attention as a natural molecule with significant affinity towards targets of potential medicinal interest, such as estrogen receptor, tyrosine kinases and topoisomerase II. The efforts in designing genistein analogs and conjugates are aimed at obtaining compounds with improved efficacy and selectivity towards selected targets; these efforts point to some specific derivatives presenting enhanced binding to known molecular targets of genistein or interacting with new ones, previously not recognized as being affected by this isoflavonoid. Special attention has been paid to the mechanisms of action of glycoconjugates. Antiproliferative activity of several genistein derivatives against cancer cell lines in vitro, including inhibition of tyrosine kinases, inhibition of topoisomerase II, destablilisation of spindle microtubules and induction of apoptosis are presented. Our results indicate that derivatization of genistein with sugars can change the mode of action of such derivative inside cancer cells. New compounds may also exhibit bimodal activity: at lower concentrations some of them act on microtubules of the mitotic spindle, whereas at higher concentrations they can additionally affect tyrosine kinases and topoisomerase II.
Session III: Molecular modeling
LARGE SCALE SEARCHING FOR EXACT TANDEM REPEATS IN GENOMES BASED ON THE BURROWS - WHEELER TRANSFORM ALGORITHM
Rafał Pokrzywa, Łukasz Olczak, Andrzej Polański
Silesian University of Technology Gliwice, Poland.
DNA tandem repeats are adjacent repeating patterns in genomic sequences. Repeating patterns, also called motifs, can be of different lengths and repetitions can involve their exact or approximate copies. Tandem repeats are important loci in DNA. They can play functional roles in genomes as parts of regulatory or promoter regions in DNA, some of tandem repeats are parts of coding regions of genes. Tandem repeats have been proven to be related to several genetically inherited diseases. Tandem repeats of motifs of different lengths are abundant in genomes, which makes them very useful as genetic markers. They are therefore used in many experimental techniques in molecular biology, for example in forensics medicine for genetic fingerprinting of individuals, in parental tests, in genomics for tagging loci in the DNA, as molecular markers for cancer. Also several population genetic studies were based on data on tandem repeats in genomes of organisms. We present a very efficient algorithm for large scale searching for exact tandem repeats in genomes. The algorithm is based on the use of the Burrows–Wheeler Transform - an efficient algorithm, which makes possible quick searches of large text files along with their compression. We present several examples of the use of our algoritm. We compare our algorithm with other algorithms from the literature. We show a study of frequencies of overlapping tandem repeats obtained with the use of our algoritm. We also present the genome - scale
alignment between genomes of Homo Sapiens and Homo Neanderthalensis,
concerning tandem repeat loci. The genome of the Homo Sapiens has
already been available for several years, while the genome of Homo
Neanderthalensis was published recently. Tandem repeat loci are, up to now,
not very well mapped between genomes. In the presented research a newly
elaborated tool, Burrows-Wheeler bases tandem repeat searches (BWtrs) is used
for searching for exact tandem repeats in the two genomes. Alignment between
STRs in the genomes is elaborated by using the dynamic programming principle,
on the basis of existing annotations of the two genomes. The data on alignment
of STRs in the two genomes are used for verification of coalescence-type models
of evolution.
GENOMIC DATA ANALYSIS WITH BOOTSTRAPPING
Krzysztof Fujarewicz
Silesian University of Technology Gliwice, Poland.
The main property of multi-scale genomic data sets is that the number of observations is much less than the number of features. This is the primary source of various problems, traps and pitfalls with genomic data analysis. To make data analysis more reliable several computational techniques are used and one of them is the bootstrap technique. It belongs to a wider class of resampling methods. In the presentation we focus on supervised data analysis: gene selection and sample classification. Four different applications of the bootstrap technique are presented: (i) assessing the accuracy of the classifier and the confidence interval, (ii) bootstrap-based feature ranking, (iii) bootstrap-based outlier detection, and (iv) stability of gene lists analysis. Assessing the accuracy of genomic classifiers leads to wide confidence intervals that usually overlap for different supervised methods of gene selection and classification. For this reason there is still no evidence which methods are predestined for genomic data analysis. Recently, so called stability of gene lists became popular. Stability of gene lists stands for the invariability of the order of selected genes with respect to the data alternation. The data may be altered for example by the bootstrap resampling. We show that accuracy assessing accompanied with the gene lists stability analysis gives more reliable evaluation of various gene selection method. As an example a gene selection based on Partial Least Squares (PLS) for multi-class problems is compared to other selection methods.
This work has been supported by the Silesian University of Technology under project BK 218/RAu1/2009.
A RELIABLE AND SIMPLE APPROACH TO FEATURE SELECTION AND INTERDEPENDENCY DISCOVERY IN SUPERVISED CLASSIFICATION
Jacek Koronacki
Institute of Computer Science, Polish Acad. Sci., Warsaw.
More often than not, rather than obtaining the best possible supervised classifier, the life scientist needs to know which features contribute best to classifying observations (samples) into distinct classes and what are the interdependencies between the features which describe the observation. To this end, in 2006, we proposed an effective method for ranking features according to their importance for classification regardless of a classifier to be used [Bioinformatics 2008, 24(1):110-117]. Later, the algorithm was extended to include the functionality of finding a cut-off between informative and non-informative features and, more importantly, we continued with a development of a methodology and an implementation of a procedure for determining interdependencies between informative features. Our approach to feature selection rests on multiple construction of tree classifiers, where each classifier is trained on a randomly chosen subset of the original samples using only a fraction of all of the observed features. Regarding interdependency discovery, we focus on identifying features that cooperate in determining that a sample belongs to a particular class. We have shown that, despite its simplicity and the use of tree classifiers, the algorithm is not biased towards features with many values (categories or levels). The algorithm’s applicability will be illustrated briefly by computational analysis of molecular interaction networks underlying change of HIV-1 resistance to selected reverse transcriptase inhibitors.
This presentation is based on the joint work of Michal Draminski, Marcin Kierczak, Alvarao Rada-Iglesias, Stefan Enroth, Claes Wadelius, Krzysztof Ginalski, Witold Rudnicki, Agnieszka Nowak-Brzeinska, Tomasz Jach, Tomasz Xieski, Jacek Koronacki and Jan Komorowski. CLC BIO-SPECIALIZED SOFTWARE SOLUTIONS FOR GENETICS AND GENOMICS
Mateusz Galuszka
Selvita, Krakow, Poland.
During the seminar we will present bioinformatics solutions from CLC bio-specialized software tools for analysis and visualization of genomic data. With Next Generation Sequencing machines, high throughput sequencing has become accesible to a very large group of researchers. However, data analysis represents a serious bottleneck in NGS pipelines. CLC Genomics solutions solve this problem and enable everyone to rapidly analyze and visualize the huge amounts of data generated by NGS machines. CLC Genomics Workbench is also the first comprehensive analysis package which can analyze and visualize data from all major NGS platforms, like SOLiD by Applied Biosystems, 454 GS flx by Roche, Genome Analyzer by Illumina and HeliScope by Helicos, as well as Sanger sequencing and one-color microarrays.CLC bio develops Next Generation Sequencing solutions through close collaboration with instrument vendors and with genomics centers worldwide. Among others, CLC bio is the only company which partners with new NGS vendors: Pacific Biosciences and Ion Torrent, to ensure the compatibility of its algorithms with data generated from new platforms. EXPLORING PATTERNS OF NON-RANDOMNESS IN HUMAN MUTATIONS
William Amos
Molecular Evolution Group, Cambridge University, Cambridge,UK.
Classical models of DNA evolution were forced to use a number of simplifying assumptions such as that mutations occur independently and at random. The publication of increasing numbers of complete genome sequences allows us to challenge these assumptions and uncover a complicated world in which randomness is anything but the rule. In this talk I present a number of analyses centred around microsatellite evolution that explore how microsatellites are born, how fast they mutate and evidence that they follow a predictable life cycle. I particularly focus on an interaction between molecular evolution and demography, showing how one can influence the other. Finally, I include examples of how, having understood one process, the patterns observed can be used to test for another. In particular I look at novel ways in which the impact of natural selection on the human genome can be inferred. EVOLUTION: A GUIDE TO PROTEIN FUNCTION AND ITS REDESIGN
Olivier Lichtarge
Cullen Foundation Professor of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA.
Protein interactions underlie all aspects of biological activity. They organize cellular components into complexes, macromolecular machines, cellular pathways and biological networks that sustain development, growth and homeostasis. Upon disruption, deregulated interactions can lead to amyloidosis, to cancer, and to many other ailments. Conversely, the targeted modification of protein interactions are an emerging frontier for therapy. One of the main challenges in designing such new therapeutic approaches lies in the identification of the key sites and key amino acids that mediate these protein interactions. Such knowledge should enable, on the one hand, better analysis of the genetic variations most likely to be causally associated with disease, thus enabling better diagnosis and targeting of molecular therapy and, on the other hand, it should enable the rational design of peptides or mutations that can modify individual links in a complex web of protein networks. We shall discuss here Evolutionary Tracing (ET), a comparative method to identify protein functional sites and protein functional residues and to guide experiments that selectively block, recode, or mimic their amino acid determinants. The heart of the approach lies in coupling closely variations in sequences with phylogenetic variations. Examples in specific prokaryotic and in eukaryotic proteins will illustrate the accuracy of this phylogenomic technique. These case studies will be complemented by large scale analyses that computationally identify the function of novel protein structures and the impact of genetic varations that different individuals may harbor. In principle, these studies suggest a scalable approach to analyze genomics data so as to extract new information on the molecular basis of function and to perturb individual links in protein networks efficiently.
Reference: Lichtarge O, Wilkins A., (2010), Evolution: a guide to perturb protein function and networks. Curr Opin Struct Biol. 20(3):351-9.
PATTERNS OF EVOLUTION OF HUMAN GENETIC DISEASE WITH IMPLICATIONS FOR DEEP SEQUENCING METHODS
Marek Kimmel
Department of Statistics, Rice University, Houston TX, USA and Systems Engeenering Group, Silesian University of Technology, Gliwice, Poland.
The two main theories concerning the genomic architecture of human genetic disease are the Common Variant Common Disease (CVCD) and the Rare Variant Common Disease (RVCD) hypotheses. The talk addresses the possible evolutionary mechanisms that lead to these two patterns, the evidence for each of them, and the consequences for gene finding. In particular, we show that population genetics simulations taking into account past demography of modern humans lead to distributions of variant frequencies, consistent with either CVCD or RVCD, depending on specific assumptions. On the other hand, it is hoped that the recently introduced deep sequencing technology in conjunction with new bioinformatics and statistical methods, will lead to breakthroughs in disease gene finding. However, particularly under RVCD, the requirements for sample size might still be formidable. The talk is illustrated with examples from the literature and author´s own studies. | |